Blat
Blat,全稱 The BLAST-Like Alignment Tool,可以稱為"類BLAST 比對工具",對於DNA序列,BLAT是用來設計尋找95%及以上相似至少40個鹼基的序列。對於蛋白序列,BLAT是用來設計尋找80%及以上相似至少20個氨基酸的序列。
Blat 的主要特點就是:速度快,共線性輸出結果簡單易讀。對於比較小的序列(如 cDNA 等)對大基因組的blat與blast比較比對,blat 無疑是首選。Blat 把相關的呈共線性的比對結果連接成為更大的 比對結果,從中也可以很容易的找到 exons 和 introns。因此,在相近物種的基因同源性分析和EST 分析中,blat 得到了廣泛的應用。Blat的比對速度之所以能比Blast快幾百倍,是因為此兩者之間的比對機制有着本質的差別。Blast是將查詢序列索引化,然后線性搜索龐大的目標數據庫,期間頻繁地訪問硬盤數據,時間和空間上的數據相關性較小;Blat則將龐大的目標數據庫索引化,然后線性搜索查詢序列,這種搜索方式在時間和空間上的數據相關性比較大。Blat將數據庫索引一次性讀入內存,可以反復地高速調用,無需訪問硬盤,占用的系統資源很少。只要索引建立,查詢序列的量越大,Blat的優勢就越明顯。
Blat is an alignment tool like BLAST, but it is structured differently. Blat produces two major classes of alignments:
- at the DNA level between two sequences that are of 95% or greater identity, but which may include large inserts.
- at the protein or translated DNA level between sequences that are of 80% or greater identity and may also include large inserts.
wget -c https://users.soe.ucsc.edu/~kent/src/blatSrc35.zip
unzip blatSrc35.zip
cd blatSrc
uname -a
export MACHTYPE="x86_64"
mkdir ~/bin/$MACHTYPE
mkdir $MACHTYPE
make >make$(date +%F).log
echo 'export PATH="/public/home/guixiang/bin/x86_64:$PATH"' >> ~/.bash_profile
基本原理: 首先blat將參考序列拆分成tiles/kmers,其拆分的方式取決於兩個參數: -tileSize and -stepSize。其中 -tileSize決定tiles/kmers的大小,一般設定范圍是:8-12,預設DNA為11,蛋白質為5;-stepSize決定tiles/kmers移動的步長。
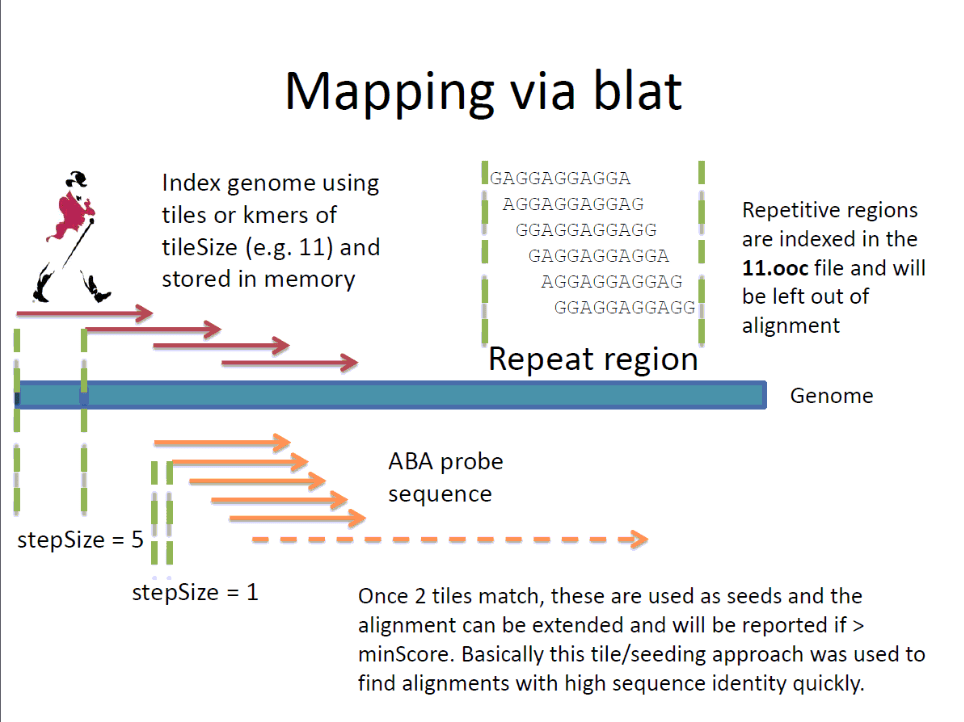
參考鏈接:Blat-The BLAST-Like Alignment Tool (詳細的使用教程)
參數詳解
#blat常見用法
#處理單個job
blat chr11.fa human/test.fa test.psl #輸出不含序列
blat chr11.fa human/test.fa -out=pslx test.pslx #輸出含序列
blat chr11.fa human/test.fa -out=blast test.blast #輸出格式同NABI的blast格式
#並行處理多個jobs
time parallel blat chr{}.fa human/human.fa test_{}.psl ::: {1..22} X Y M
#blat參數
#用法:blat database query [-ooc=11.ooc] output.psl
#database 輸入文件必須是其中一種類型:a .fa , .nib or .2bit file
#query 輸入文件必須是其中一種類型:a .fa , .nib or .2bit file
#output.psl 輸出文件
#-t=type 數據庫類型,可選項: dna/prot/dnax
#-q=type 查詢序列的類型,可選項:dna/prot/dnax/rnax
#-prot 等同於 -t=prot -q=prot
#-ooc=N.ooc Use overused tile file N.ooc. N should correspond to the tileSize
#-tileSize=N 設定tiles/kmers的大小
#-stepSize=N 設定tiles/kmers在比對時移動的步長,即兩個相鄰tiles/kmers之間的距離,預設值是tileSize
#-oneOff=N 如果設定為 1 ,則表示在比對到tile上允許有一個錯配鹼基(mismatch),預設值是0
#-minMatch=N 設定至少匹配的tile的個數,一般設置值的范圍是2-4,通常核苷酸的預設值為2,蛋白質的預設值為1
#-minScore=N 設定最小分值。 由於indel通常會對序列的功能產生影響,所以空位在比對過程中總是對應於一個負分,也就是所謂的空位罰分(Gap penalty)。根據打分機制,這個分值等於匹配鹼基分值減去替換分值(mismatch)和空位罰分。預設值為30
#-minIdentity=N 設置序列相似度(sequence identity)最小百分比。通常核苷酸(nucleotide searches)預設值為90,蛋白質和翻譯蛋白(protein or translated protein searches)預設值為25
#-maxGap=N 在一定長度序列中,設定兩個tiles/kmers之間的允許最大的空位(gap)大小。通常設定范圍是0-3,預設值為2,且僅在minMatch > 1時搭配使用
#-noHead 抑制.psl頭文件的輸出,內容全部均是以制表符為分隔符的文件
#-makeOoc=N.ooc Make overused tile file. Target needs to be complete genome.
#-repMatch=N 在一段序列被標記為overused之前,設定允許tiles/kmers重復次數。如果超過設定值,該tiles/kmers將會被標記為overused。通常當tileSize設定為12時,repMatch則設定為256;當tileSize設定為11時,repMatch則設定為1024;當tileSize設定為10時,repMatch則設定為4096。
#-mask=type Mask out repeats. Alignments won't be started in masked region but may extend through it in nucleotide searches. Masked areas are ignored entirely in protein or translated searches. Types are
#lower - mask out lower cased sequence
#upper - mask out upper cased sequence
#out - mask according to database.out RepeatMasker .out file
#file.out - mask database according to RepeatMasker file.out
#-qMask=type Mask out repeats in query sequence. 類型選擇與參數-mask相同
#-repeats=type 類型選擇與參數-mask相同。無論如何重復鹼基不會被掩蓋(masked),但是在匹配重復區域時將會在psl輸出文件中會單獨展示其匹配結果,即與其他區域的匹配結果是分開的。
#-minRepDivergence=NN - minimum percent divergence of repeats to allow them to be unmasked. Default is 15. Only relevant for masking using RepeatMasker .out files.
#-dots=N 每N個序列就輸出一個點,用於展示程序運行的進度
#-trimT 剪切首部的poly-T
#-noTrimA 不剪切尾部的poly-A
#-trimHardA 從psl輸出文件中的qSize和alignments中移除poly-A尾巴
#-fastMap 快速的DNA/DNA remapping,要求查詢序列長度不超過5000、高相似度和不進行內含子的比對
#-out=type 輸出文件格式,格式如下:
# psl - Default. Tab separated format, no sequence
# pslx - Tab separated format with sequence
# axt - blastz-associated axt format
# maf - multiz-associated maf format
# sim4 - similar to sim4 format
# wublast - similar to wublast format
# blast - similar to NCBI blast format
# blast8- NCBI blast tabular format
# blast9 - NCBI blast tabular format with comments
#-fine 對於高質量的mRNAs搜索small initial和terminal exons更為嚴苛。此選項不推薦應用於ESTs
#For high quality mRNAs look harder for small initial and terminal exons.
#-maxIntron=N 設定內含子最大的序列長度. Default is 750000
#-extendThroughN - 允許序列的比對可以從大段N區域延伸
GNU Parallel
#安裝編譯
wget ftp://ftp.gnu.org/gnu/parallel/parallel-20170822.tar.bz2
tar -jxvf parallel-20170822.tar.bz2
cd parallel-20170822/
cat README
./configure && make && sudo make install
#使用
#parallel教程: http://www.gnu.org/software/parallel/parallel_tutorial.html
#parallel中文版教程: http://my.oschina.net/enyo/blog/271612
#parallel與其他Linux命令的搭配使用: http://www.vaikan.com/use-multiple-cpu-cores-with-your-linux-commands/