分析帶UMI標簽的測序數據
分析帶UMI標簽的測序數據
檢測癌組織的低頻突變,為了提高檢測低頻突變的靈敏度,往往進行高深度的測序。但樣本之間存在交叉污染,測序有存在一定概率的錯誤,這些因素會導致高深度測序過程中將假陽性的信號放到,得到假陽性的結果。解決交叉污染的方法,有公司比如IDT采用唯一配對的樣本index,只有配對的index中的reads才屬於特定樣本。解決測序錯誤的方法,研究人員在建庫的時候,先對分子加上UMI鹼基,unique molecular identifier -> UMI,然后根據來源於同一個分子的測序數據進行測序錯誤修正,得到正確的分子序列。兩種方法結合可以減少交叉污染提高准確性(https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5759201/)。
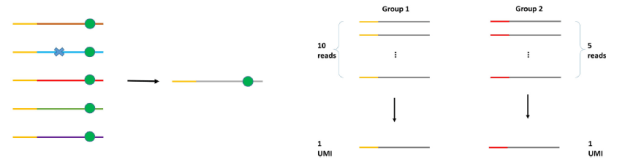
如圖中所示,左側一個分子被測了5次,其中第二次有一個測序錯誤,但該錯誤並沒有在每個測序數據中出現,所以在后續合成一個分子的時候,測序錯誤被修正,只保留了真正的突變。(https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5852328/)
常規的腫瘤配對測序分析,或者遺傳性突變位點的分析,並不需要UMI信息,所以包含UMI的數據分析是需要不一樣的分析流程來得到准確的分析結果,其中包括提取UMI分子標簽,合並來自同一個分子的測序reads,低頻突變檢測而非胚系突變檢測等。
大致流程為:
-----prepare analysis ready BAM file------ | FASTQ | ↓ | uBAM | ↓ Extract UMI | uBAM | ↓ Align uBAM and merge | BAM -----call consensus------ | ↓ Group Reads By Umi | BAM | ↓ Group Reads By Umi | BAM | ↓ Call Molecular Consensus Reads | uBAM | ↓ Align uBAM and merge | BAM | ↓ Filter Consensus Reads | BAM | ↓ Clip | BAM -----Vardict------ | ↓ Call | VCF
一,得到包含UMI分子標簽信息的BAM文件
UMI信息,應該從fastq中的配對的reads中提取,但fastq不能存儲更多的信息,所以需要先將fastq轉成uBAM文件,提取uBAM文件中的UMI分子標簽信息,將該信息通過RX標簽寫入uBAM文件中,通過uBAM和BAM文件合並,把RX信息合並到比對到的BAM文件中進行下一步分析。
1)生成uBAM
java -Xmx8G -jar picard.jar FastqToSam \
FASTQ=$fq1 \
FASTQ2=$fq2 \
OUTPUT=test.uBAM \
READ_GROUP_NAME=test \
SAMPLE_NAME=test \
LIBRARY_NAME=test \
PLATFORM_UNIT=HiseqX10 PLATFORM=illumina \
RUN_DATE=`date --iso-8601=seconds`
2)提取UMI信息
java -jar fgbio.jar ExtractUmisFromBAM \
--input=test.uBAM \
--output=test.umi.uBAM \
--read-structure=2M148T 2M148T \
--single-tag=RX \
--molecular-index-tags=ZA ZB
此時,uBAM文件中RX標簽記錄着UMI的信息,2M148T表示前兩個鹼基是UMI分子標簽,148個template 測序序列,配對read在uBAM文件中有如下信息
R1----ZA:Z:TC ZB:Z:TA RG:Z:test RX:Z:TC-TA R2----ZA:Z:TC ZB:Z:TA RG:Z:test RX:Z:TC-TA
3)比對uBAM文件中的reads
samtools fastq test.umi.uBAM | bwa mem -t 8 -p reference.fa /dev/stdin | samtools view -b > test.umi.BAM
4)uBAM和BAM合並
java -Xmx8G -jar picard.jar MergeBAMAlignment R=reference.fa \
UNMAPPED_BAM=test.umi.uBAM \
ALIGNED_BAM=test.umi.BAM \
O=test.umi.merged.BAM \
CREATE_INDEX=true \
MAX_GAPS=-1 \
ALIGNER_PROPER_PAIR_FLAGS=true \
VALIDATION_STRINGENCY=SILENT \
SO=coordinate \
ATTRIBUTES_TO_RETAIN=XS
通過合並,得到一個包含各種信息包括RX tag等的BAM文件,該文件用於下一步call consensus read,因為將來源於同一個分子的read合並成一個consensus read,所以在得到BAM文件之后,沒有進行mark duplication。
二,Call Consensus Reads
1)Group Reads By Umi
該步會生成一個包含MI tag的文件,存儲每個read的最初分子的ID,低質量的read應該過濾掉,因為低比對質量的read上mismatch較多,容易造成假陽性。
java -jar fgbio.jar GroupReadsByUmi \
--input=test.umi.merged.BAM \
--output=test.umi.group.BAM \
--strategy=paired --min-map-q=20 --edits=1 --raw-tag=RX
2)Call Molecular Consensus Reads
根據UMI分子標簽的信息和Read1、Read2的位置,從一組read中識別出最初的 molecular 分子序列。min-reads是必填的,如果測序深度較低,單個read也可以用來call consensus,此時設置1,如果深度較高可以設置2或者更高,但1是最安全的,因為后續可以基於此參數進一步過濾。但同時該值的設置顯著影響效率,此時我們設置1。此時生成的文件是uBAM格式的,需要進一步比對
java -jar fgbio.jar CallMolecularConsensusReads \
--min-reads=1 \
--min-input-base-quality=20 \
--input=test.umi.group.BAM \
--output=test.consensus.uBAM
3)比對uBAM文件中的reads
samtools fastq $dir/$smp.callconsensus.BAM | bwa mem -t 8 -p reference.fa /dev/stdin | samtools view -b - | test.consensus.BAM
4)uBAM和BAM合並
java -Xmx8G -jar picard.jar MergeBAMAlignment R=reference.fa \
UNMAPPED_BAM=test.consensus.uBAM \
ALIGNED_BAM=test.consensus.BAM \
O=test.consensus.merge.BAM \
CREATE_INDEX=true \
MAX_GAPS=-1 \
ALIGNER_PROPER_PAIR_FLAGS=true \
VALIDATION_STRINGENCY=SILENT \
SO=coordinate \
ATTRIBUTES_TO_RETAIN=XS
4)Filter Consensus Reads
java -jar fgbio.jar FilterConsensusReads \
--input=test.consensus.merge.BAM \
--output=test.consensus.merge.filter.BAM \
--ref=reference --min-reads=2 \
--max-read-error-rate=0.05 \
--max-base-error-rate=0.1 \
--min-base-quality=30 \
--max-no-call-fraction=0.20
5)Clip
來源於同一個template的read,若果有重疊,重疊部分的突變其實應該只計一次,所以要clip一下read。
java -jar fgbio.jar ClipBAM \
--input=test.consensus.merge.filter.BAM \
--output=test.consensus.merge.filter.clip.BAM \
--ref=$ref --soft-clip=false --clip-overlapping-reads=true
三、Variant Call
AF_THR="0.01"
VarDict/bin/VarDict -G reference.fa -f $AF_THR -N test \
-b test.consensus.merge.filter.BAM \
-z -c 1 -S 2 -E 3 -g 4 -th 4 target.bed | \
VarDictJava/VarDict/teststrandbias.R | \
VarDictJava/VarDict/var2vcf_valid.pl -N test -E -f $AF_THR > test.vcf
參考:
https://github.com/fulcrumgenomics/fgbio
https://gatkforums.broadinstitute.org/gatk/discussion/6484/how-to-generate-an-unmapped-BAM-from-fastq-or-aligned-BAM
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5759201/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5852328/
https://www.ncbi.nlm.nih.gov/pubmed/28100584
#####################################################################
#版權所有 轉載請告知 版權歸作者所有 如有侵權 一經發現 必將追究其法律責任
#Author: Jason
#####################################################################