文章目錄
- RNA-seq 數據分析流程
-
- 相關軟件安裝
- 下載數據
-
- sra轉fastq格式
- 數據質控
-
- 數據質控,過濾低質量reads,去接頭
- 比對
-
- 首先下載參考基因組及注釋文件,建立索引
- 比對
- sam文件轉bam
- 為bam文件建立索引
- reads的比對情況統計
- 計數 counts
- 差異基因分析
RNA-seq 數據分析流程
相關軟件安裝
可以安裝 conda,在后續其他軟件安裝時非常好用。可自行百度進行安裝
可根據文獻調研,轉錄組數據分析所需軟件列表:
質控
fastqc , multiqc, trimmomatic, cutadapt ,trim-galore
比對
star, hisat2, bowtie2, tophat, bwa, subread
計數
htseq, bedtools, deeptools, salmon
在安裝之前可以先 search一下安裝包是否存在
# conda search packagename
conda install -y sra-tools
conda install -y trimmomatic
conda install -y cutadapt multiqc
conda install -y trim-galore
conda install -y star hisat2 bowtie2
conda install -y subread tophat htseq bedtools deeptools
conda install -y salmon
軟件安裝時,可以使用conda一步安裝,也可以自行百度,下載源碼,解壓后添加環境變量,使用。
但是在使用sra-tookit時出現了問題,因為已經提前用conda安裝好了sra-
tools,后面我在網頁下載解壓了sratoolkit,並且在環境變量中添加好了,導致兩個軟件版本不合,使用prefetch下載網頁數據時總是報錯。所以需要卸載其中一種,保持版本一致。
自行安裝sratoolkit可參考: sratoolkit安裝
下載數據
進入網頁,得到SRR_Acc_list.txt
SRR957677
SRR957678
SRR957679
SRR957680
批量下載
wkd=/home/meiling/baiduyundisk/RNA-seq #設置工作目錄
cat SRR_Acc_List.txt | while read id; do (nohup prefetch ${id} &);done
#ps -ef | grep prefetch | awk '{print $2}' | while read id; do kill ${id}; done #在內地下載速度很慢,所以我殺掉這些下載進程
當顯示所有下載都sucessful時,可以進行下一步
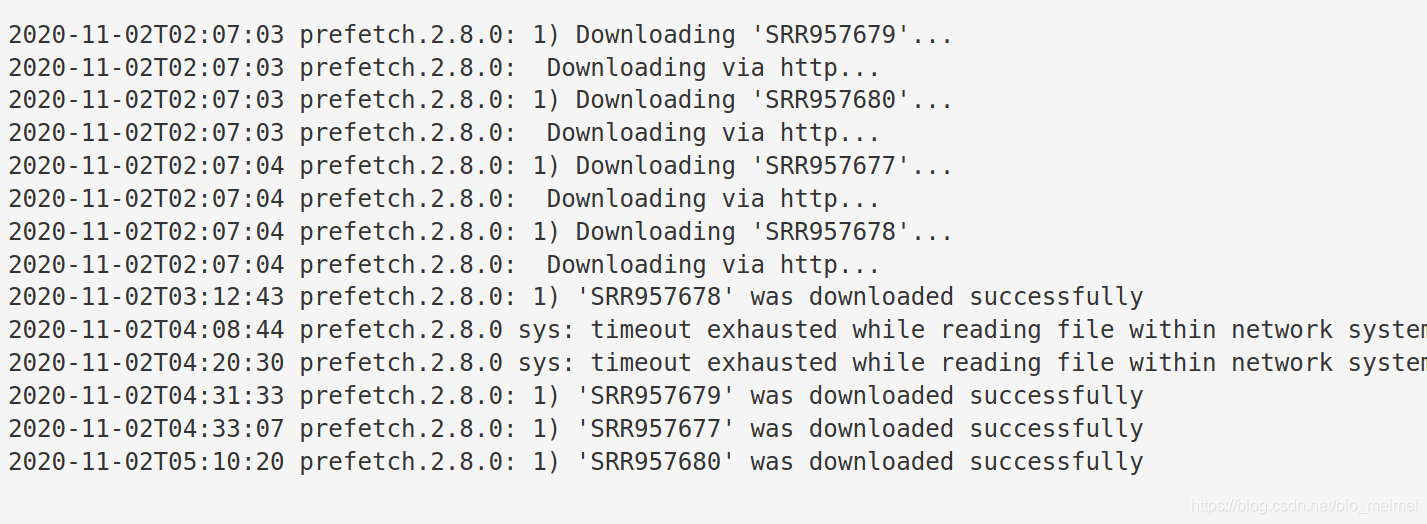
下載完成之后,文件會自動保存在目錄:~/ncbi/public/sra/
mv ~/ncbi/public/sra/*.sra $wkd/rawdata
sra轉fastq格式
使用fastq-dump
wkd=/home/meiling/baiduyundisk/RNA-seq #設置工作目錄
for i in $wkd/rawdata/*sra
do
echo $i
fastq-dump --split-3 --skip-technical --clip --gzip $i ## 批量轉換
done
原始數據如果是雙端測序結果,fastq-dump配合–split-3參數,一個樣本被拆分成兩個fastq文件,如果是單端測序,就只生成一個文件
數據質控
fastqc生成質控報告,multiqc將各個樣本的質控報告整合為一個。
wkd=/home/meiling/baiduyundisk/RNA-seq #設置工作目錄
ls $wkd/rawdata/*gz | xargs fastqc -t 2
multiqc ./
得到結果如下:
├── [4.0K] multiqc_data
│ ├── [2.1M] multiqc_data.json
│ ├── [6.8K] multiqc_fastqc.txt
│ ├── [2.2K] multiqc_general_stats.txt
│ ├── [ 16K] multiqc.log
│ └── [3.4K] multiqc_sources.txt
├── [1.5M] multiqc_report.html
├── [236K] SRR1039508_1_fastqc.html
├── [279K] SRR1039508_1_fastqc.zip
├── [238K] SRR1039508_2_fastqc.html
├── [286K] SRR1039508_2_fastqc.zip
每個id_fastqc.html都是一個質量報告,multiqc_report.html是所有樣本的整合報告
數據質控,過濾低質量reads,去接頭
如果雙端測序,運行一下代碼,得到兩列數據,config文件
mkdir $wkd/clean
cd $wkd/clean
ls /home/jmzeng/project/airway/raw/*_1.fastq.gz >1
ls /home/jmzeng/project/airway/raw/*_2.fastq.gz >2
paste 1 2 > config
並打開qc.sh,寫入一下內容:
bin_trim_galore=trim_galore
dir='/home/jmzeng/project/airway/clean'
cat $1 |while read id
do
arr=(${id})
fq1=${arr[0]}
fq2=${arr[1]}
$bin_trim_galore -q 25 --phred33 --length 36 --stringency 3 --paired -o $dir $fq1 $fq2
done
單端測序:
wkd=/home/meiling/baiduyundisk/RNA-seq #設置工作目錄
mkdir $wkd/cleandata
cd $wkd/cleandata
ls /home/meiling/baiduyundisk/RNA-seq/rawdata/*.fastq.gz >config
#ls /home/jmzeng/project/airway/raw/*_2.fastq.gz >2
#paste 1 2 > config
qc.sh
wkd=/home/meiling/baiduyundisk/RNA-seq/cleandata #設置工作目錄
cd $wkd
cat config |while read id
do
arr=(${id})
fq1=${arr[0]}
# fq2=${arr[1]}
trim_galore -q 25 --phred33 --length 36 --stringency 3 -o $wkd $fq1
done
其他軟件,對於單端測序和雙端測序的不同用法,可以自行百度具體軟件用法,都有解釋,參考官方參數即可。
比對
首先下載參考基因組及注釋文件,建立索引
基因組相關內容大家可以自行百度,但是參考基因組和注釋文件一定要對應在同一個網站,同一個版本。可以下載的網站有UCSC,NCBI,ENSEMBEL
我是在ENSEMBEL上面下載的,一般選擇primary參考基因組,注釋文件選擇scaff
復制鏈接地址后,axel 下載,非常快。下載后解壓。
axel -n 20 ftp://ftp.ensembl.org/pub/release-101/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
axel -n 20 ftp://ftp.ensembl.org/pub/release-101/gtf/homo_sapiens/Homo_sapiens.GRCh38.101.chr_patch_hapl_scaff.gtf.gz
ls
#Homo_sapiens.GRCh38.101.chr_patch_hapl_scaff.gtf.gz
#Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
##解壓
gzip -d Homo_sapiens.GRCh38.101.chr_patch_hapl_scaff.gtf.gz
gzip -d Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
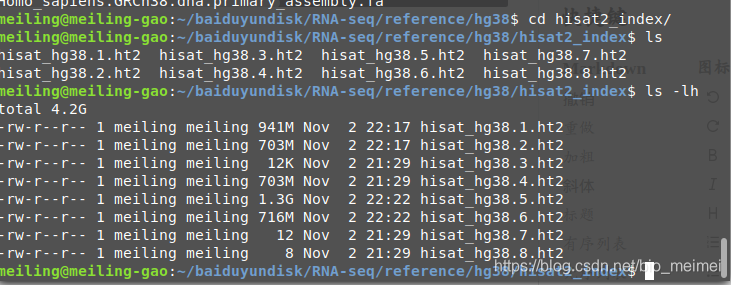
ls -lh
total 4.3G
drwxr-xr-x 2 meiling meiling 4.0K Nov 2 21:26 hisat2_index
-rw-r--r-- 1 meiling meiling 1.3G Nov 1 20:48 Homo_sapiens.GRCh38.101.chr_patch_hapl_scaff.gtf
-rw-r--r-- 1 meiling meiling 3.0G Nov 1 20:50 Homo_sapiens.GRCh38.dna.primary_assembly.fa
##建立索引
hisat2-build Homo_sapiens.GRCh38.dna.primary_assembly.fa hisat2_index/hisat_hg38
比對
star, hisat2, bowtie2, tophat, bwa, subread都是可以用於比到的軟件
批量比對:
wkd=/home/meiling/baiduyundisk/RNA-seq #設置工作目錄
cd $wkd/cleandata
ls *gz|cut -d"_" -f 1 |sort -u |while read id;do
##單端測序
ls -lh ${id}_trimmed.fq.gz
hisat2 -q -p 10 -x $wkd/reference/hg38/hisat2_index/hisat_hg38 -U ${id}_trimmed.fq.gz -S $wkd/align/${id}.hisat.sam
##雙端測序
#ls -lh ${id}_1_val_1.fq.gz ${id}_2_val_2.fq.gz
#hisat2 -p 10 -x $wkd/reference/hg38/hisat2_index/hisat_hg38 -1 ${id}_1_val_1.fq.gz -2 ${id}_2_val_2.fq.gz -S ${id}.hisat.sam
# subjunc -T 5 -i /public/reference/index/subread/hg38 -r ${id}_1_val_1.fq.gz -R ${id}_2_val_2.fq.gz -o ${id}.subjunc.sam
# bowtie2 -p 10 -x /public/reference/index/bowtie/hg38 -1 ${id}_1_val_1.fq.gz -2 ${id}_2_val_2.fq.gz -S ${id}.bowtie.sam
# bwa mem -t 5 -M /public/reference/index/bwa/hg38 ${id}_1_val_1.fq.gz ${id}_2_val_2.fq.gz > ${id}.bwa.sam
done
這里是演示多個比對工具,但事實上,對RNA-seq數據來說,不要使用bwa和bowtie這樣的軟件,它需要的是能進行跨越內含子比對的工具。
比對結果輸出:
bash align.sh > align.log 2>&1
##打開align.log
###單端測序比對結果輸出
-rw-r--r-- 1 meiling meiling 984M Nov 2 16:33 SRR957677_trimmed.fq.gz
File "/opt/software/hisat2-2.2.0/hisat2_read_statistics.py", line 182
length_map = sorted(length_map.iteritems(), key=lambda (k,v):(v,k), reverse=True)
^
SyntaxError: invalid syntax
20498068 reads; of these:
20498068 (100.00%) were unpaired; of these:
689901 (3.37%) aligned 0 times
17438548 (85.07%) aligned exactly 1 time
2369619 (11.56%) aligned >1 times
96.63% overall alignment rate

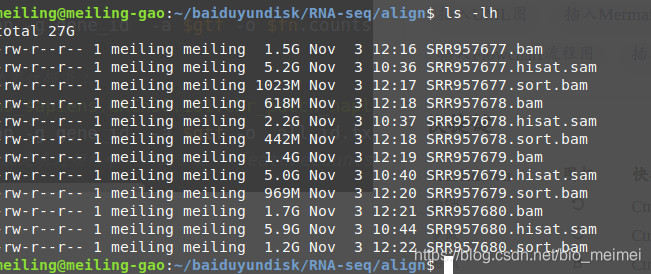
sam文件轉bam
wkd=/home/meiling/baiduyundisk/RNA-seq #設置工作目錄
cd $wkd/align
ls *.sam|cut -d"." -f 1 |while read id ;do
samtools view -@ 8 -S {$id}.sam -1b -o {$id}.bam
samtools sort -@ 8 -l 5 -o {$id}.sort.bam {$id}.bam
done
#rm *.sam
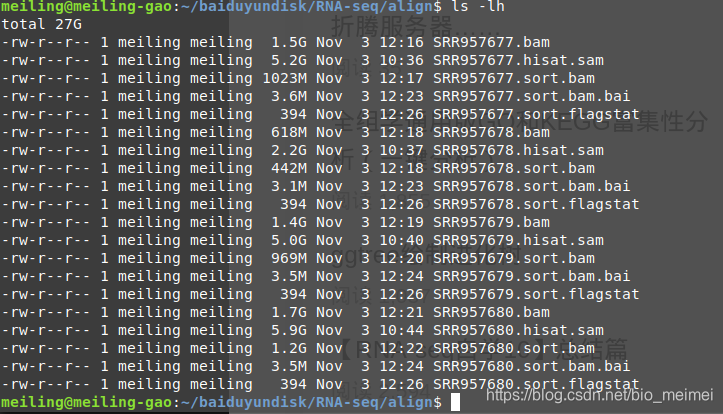
為bam文件建立索引
ls *.sort.bam |xargs -i samtools index {}
reads的比對情況統計
ls *.sort.bam |while read id ;do ( samtools flagstat -@ 1 $id > $(basename ${id} ".bam").flagstat );done
計數 counts
首先安裝featurecounts,這個軟件已經整合到subread里面了
axel -n 20 https://udomain.dl.sourceforge.net/project/subread/subread-2.0.1/subread-2.0.1-Linux-x86_64.tar.gz
tar -zxvf subread-2.0.1-Linux-x86_64.tar.gz
cd subread-2.0.1-Linux-x86_64/
cd bin
pwd
#添加環境變量
計數
wkd=/home/meiling/baiduyundisk/RNA-seq #設置工作目錄
cd $wkd/align
# 如果一個個樣本單獨計數,輸出多個文件使用代碼是:
for fn in {508..523}
do
featureCounts -T 5 -p -t exon -g gene_id -a $gtf -o $fn.counts.txt SRR1039$fn.bam
done
# 如果是批量樣本的bam進行計數,使用代碼是:
wkd=/home/meiling/baiduyundisk/RNA-seq #設置工作目錄
cd $wkd/align
# 如果一個個樣本單獨計數,輸出多個文件使用代碼是:
# for fn in {508..523}
# do
# featureCounts -T 5 -p -t exon -g gene_id -a /public/reference/gtf/gencode/gencode.v25.annotation.gtf.gz -o $fn.counts.txt SRR1039$fn.bam
# done
# 運行featureCounts對所有樣本進行基因水平定量
# 如果是批量樣本的bam進行計數,使用代碼是:
# 設置featureCounts所需要的gtf文件位置(注釋文件)
gtf="$wkd/reference/hg38/Homo_sapiens.GRCh38.101.chr_patch_hapl_scaff.gtf.gz"
featureCounts -T 5 -t exon -g gene_id -a $gtf -o $wkd/counts/all.id.txt *.sort.bam 1>counts.id.log 2>&1
#解釋:-T 1:線程數為1;-p:表示數據為paired-end,雙末端測序數據;-t exon表示 feature名稱為exon;-g gene_id表示meta-feature名稱為gene_id(ensembl名稱);-a $gtf 表示輸入的GTF基因組注釋文件;-o all.id.txt 設置輸出文件類型;*.sort.bam是被分析的bam文件。featureCounts支持通配符*
# 這樣得到的 all.id.txt 文件就是表達矩陣啦,但是,這個 featureCounts有非常多的參數可以調整。
cd $wkd/counts
# 使用multiqc對featureCounts統計結果進行可視化。
multiqc all.id.txt.summary
# 只保留all.id.txt文件的【基因名】和【樣本counts】
cat all.id.txt | cut -f 1,7 |grep -v '^#' > counts.txt
運行結束:
得到以下文件:

# Program:featureCounts v2.0.1; Command:"featureCounts" "-T" "5" "-t" "exon" "-g" "gene_id" "-a" "/home/meiling/baiduyundisk/RNA-seq/reference/hg38/Homo_sapiens.GRCh38.101.chr_patch_hapl_scaff.gtf.gz" "-o" "/home/meiling/baiduyundisk/RNA-seq/counts/all.id.txt" "SRR957677.sort.bam" "SRR957678.sort.bam" "SRR957679.sort.bam" "SRR957680.sort.bam"
Geneid Chr Start End Strand Length SRR957677.sort.bam SRR957678.sort.bam SRR957679.sort.bam SRR957680.sort.bam
ENSG00000223972 1;1;1;1;1;1;1;1;1 11869;12010;12179;12613;12613;12975;13221;13221;13453 12227;12057;12227;12721;12697;13052;13374;14409;13670 +;+;+;+;+;+;+;+;+ 1735 0 0 0 0
ENSG00000227232 1;1;1;1;1;1;1;1;1;1;1 14404;15005;15796;16607;16858;17233;17606;17915;18268;24738;29534 14501;15038;15947;16765;17055;17368;17742;18061;18366;24891;29570 -;-;-;-;-;-;-;-;-;-;- 1351 7 1 2 3
ENSG00000278267 1 17369 17436 - 68 0 0 0 0
ENSG00000243485 1;1;1;1;1 29554;30267;30564;30976;30976 30039;30667;30667;31109;31097 +;+;+;+;+ 1021 0 0 0 0
ENSG00000284332 1 30366 30503 + 138 0 0 0 0
ENSG00000237613 1;1;1;1;1 34554;35245;35277;35721;35721 35174;35481;35481;36081;36073 -;-;-;-;- 1219 0 0 0 0
ENSG00000268020 1 52473 53312 + 840 0 0 0 0
ENSG00000240361 1;1;1;1 57598;58700;62916;62949 57653;58856;64116;63887 +;+;+;+1414 0 0 0 0
可以單個樣本計數,之后在進行合並,使用Python腳本或者R語言merge
具體可以參考這里
或者直接批量比對,輸出的all.id.txt即為合並后的表達矩陣。可以在這里選擇注釋,或者找完差異基因后在進行注釋。
差異基因分析
后續需要在R里面進行差異基因分析,DESeq和edger里面自帶標准化的函數,后續還可以進行KEGG,GSEA,GO富集分析
