本文出自於http://www.bioinfo-scrounger.com
之前大致了解了些免疫療法的相關內容,最近一篇文章深度長文:一文盡覽PD-1/PD-L1/CTLA-4腫瘤免疫治療分子標志物大全概括性的講述了一些現在免疫療法中生物標志物的相關內容,值得仔細看一看
先總結下其中一些感興趣的內容:
最近幾年PD-1/PD-L1在一些腫瘤免疫治療中的突破性的進展令人鼓舞,這也推進了人們對於免疫治療相關的生物標志物的研究:如常見生化反應手段的PD-L1表達,腫瘤浸潤淋巴細胞(TIL),還有聽的比較多的微衛星不穩定性(MSI)和錯配修復缺陷(dMMR),以及最近研究火熱的腫瘤突變負荷(TMB)
從測序的角度來說,可以研究分析的生物標志物(上述提到的)有MSI/dMMR/TMB
近幾年不斷研究報道TMB與PD-1/PD-L1抑制劑的療效高度相關,而以TMB為標志物的臨床研究大多數都順利達到終點,幾乎沒有失敗過。這讓一些腫瘤患者可以通過TMB標志物還對免疫療法的療效進行一定程度的預測
TMB(Tumor mutation burden)既然這么熱,那么我需要知道TMB是如何計算的:
TMB的定義通常是基因組中每1Mb蛋白編碼區的平均突變個數。所以我們可以通過WES全外顯子測序,通過call somatic variant(去除胚系DNA變異),然后除以人類蛋白編碼區長度MB即可(我覺得。。。)
這篇文章剛好也提到了之前我一朋友推薦我看的一篇文獻:Blood-based tumor mutational burden as
a predictor of clinical benefit in non-small-cell
lung cancer patients treated with atezolizumab,其中提到了bTMB(blood TMB)這個概念,也就是不檢測常規的腫瘤組織樣的tTMB,而是血液樣本的bTMB,從而避免組織樣本的一些局限性
文獻中bTMB用的篩選指標是:SNVs with ≥0.5% frequency were analyzed, and germline
variants and oncogenic driver mutations were removed
而文中所說常規的組織樣本的篩選標准:tTMB
was defined as the number of somatic, coding, SNVs and indels detected at an
allele frequency of ≥5% after the removal of known and probably oncogenic
driver events and germline SNPs, as previously described
要想獲得靠譜的bTMB,在測序中一定要使用雙端樣本index (即UDI,針對Illumina平台),否則你將無法排除Index Misassignment帶來的樣本間污染導致的大量假陽性超低頻突變。同時,還需要使用UMI分子標簽方法,以排除PCR, 和測序過程中帶來的大量錯誤
說了一些基礎概念后,那么怎么計算TMB呢;如果你有突變分析(可以是WES的,也可以是大Panel的)后的MAF格式的文件,那么只需要按照文獻中所說的,先過濾掉一些突變位點,然后統計數目再除以蛋白編碼區域的長度即可
那么什么是MAF(Mutation Annotation Format ),可以看下這個鏈接https://docs.gdc.cancer.gov/Data/File_Formats/MAF_Format/,里面除了說了什么MAF外,還對MAF格式的每個列名做了解釋說明,一般來說我們從TCGA下載是突變文件都是MAF格式的
如果是VCF格式的突變分析結果,那么可以用vcf2maf工具從vcf轉化為MAF格式,至於為什么我想說要轉為MAF格式呢,因為有個maftools這個R包,其基礎MAF格式可以做很多分析以及可視化工作,特別方便!
maftools手冊中提到,如果是ANNOVAR注釋結果的文件,可以用其
annovarToMaf將annovar outputs 轉化為MAF文件
如果你是其他公共數據,則可以通過人工處理將必要數據整理成MAF格式的文件,maftools需要的必要列有:Hugo_Symbol, Chromosome, Start_Position, End_Position, Reference_Allele, Tumor_Seq_Allele2, Variant_Classification, Variant_Type and Tumor_Sample_Barcode,另外還可以額外再加上VAF (Variant Allele Frequecy)列和amino acid change information
我們可以先用maftools包的測試數據先嘗試一遍這個R包的關鍵幾個用法(比較常見的),如:
laml.maf = system.file('extdata', 'tcga_laml.maf.gz', package = 'maftools') #path to TCGA LAML MAF file laml.clin = system.file('extdata', 'tcga_laml_annot.tsv', package = 'maftools') # clinical information containing survival information and histology. This is optional laml = read.maf(maf = laml.maf, clinicalData = laml.clin)然后看下laml這個對象存儲了哪個內容:
getSampleSummary(laml) getGeneSummary(laml) getClinicalData(laml) getFields(laml)接下來可以看看maftools根據所提供的MAF文件能做哪些分析以及可視化工作了,先是一個summary整合圖
plotmafSummary(maf = laml, rmOutlier = TRUE, addStat = 'median', dashboard = TRUE, titvRaw = FALSE)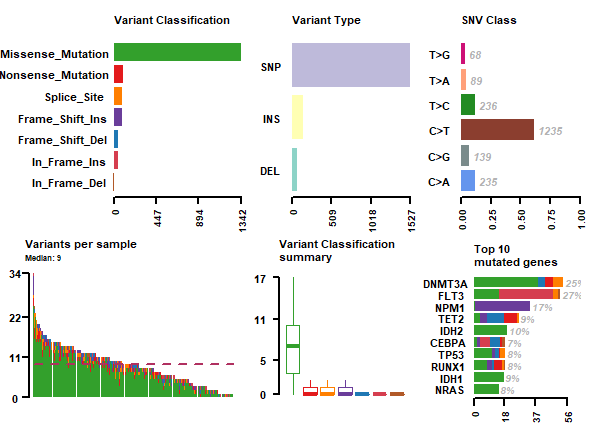
接下來是可能是最為常見的oncoplots圖,其oncoplot函數主要是在ComplexHeatmap包的OncoPrint函數基礎上做了一定的修改,當然圖形展現形式還是一樣的,主要展示每個樣本在一些基因上突變情況,可以簡單的看一下
oncoplot(maf = laml, top = 10, fontSize = 12)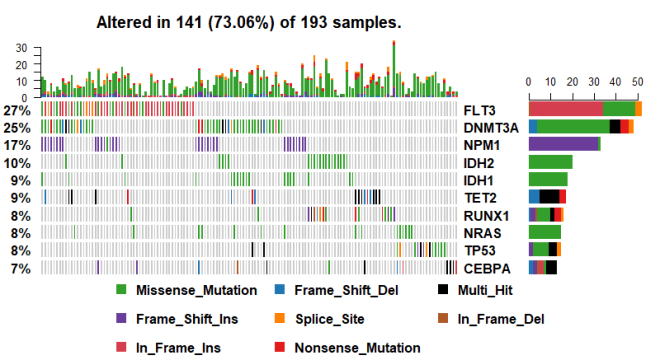
如果你的MAF文件有VAF信息的話,可以簡單畫個箱線圖看看所有樣本在不同gene上的VAF分布,vafCol就是指定MAF列中的VAF的列名
plotVaf(maf = laml, vafCol = 'i_TumorVAF_WU')
接下來則是我覺得比較有意思的:Detecting cancer driver genes based on positional clustering,這個方法是通過突變位點的位置信息,基於oncodriveCLUST算法來識別cancer genes
laml.sig = oncodrive(maf = laml, AACol = 'Protein_Change', minMut = 5, pvalMethod = 'zscore') head(laml.sig) plotOncodrive(res = laml.sig, fdrCutOff = 0.1, useFraction = TRUE)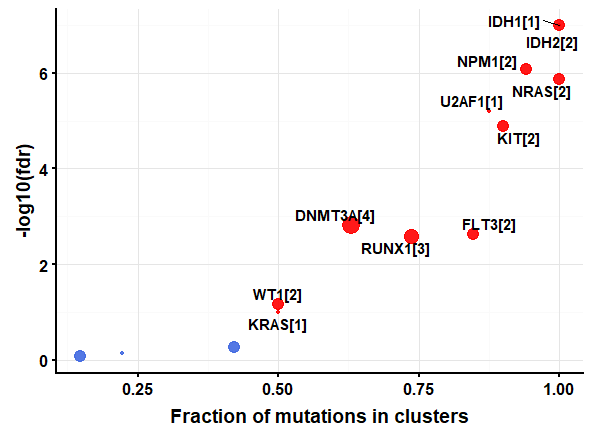
從上圖可看出,縱坐標越大表示FDR越小可行度越高;而橫坐標在表示那些突變位點聚到一個cluster的比例,而點的大小(中括號中的數字)則表示cluster的數目,所以 IDH1 has a single cluster and all of the 18 mutations are accumulated within that cluster
最后則是這篇筆記的重點內容了,之前有篇博文Mutant-allele tumor heterogeneity(MATH)提到過MATH這個生物標志物,一些文獻也對其在不同腫瘤中的預測效果進行了研究,剛好生信技能樹的一篇軟文使用Mutant-allele tumor heterogeneity(MATH)算法評估腫瘤異質性中提到使用maftools可以計算MATH
maftools的inferHeterogeneity函數可以對MAF格式的數據中的VAF列進行計算MATH,其算法跟傳統的MATH不完全一樣,其還加入了聚類算法(using mclust),相當於除去了一些outline的VAF值后再計算MATH,用測試數據的話方法如下:
tcga.ab.2972.het = inferHeterogeneity(maf = laml, tsb = 'TCGA-AB-2972', vafCol = 'i_TumorVAF_WU') > unique(tcga.ab.2972.het$clusterData$MATH) [1] 11.62157我們也可以拿TCGA的MAF突變數據嘗試下,先從TCGA官網上下載相關數據,比如我選擇BRCA-mutect2的somatic數據TCGA.BRCA.mutect.somatic.maf(名字原來不是這個,我改了下)
然后就是用maftools導入R中,看下TCGA的MAF文件有多少(很多列。。)
data <- read.maf(maf = "TCGA.BRCA.mutect.somatic.maf") getFields(data)接着因為要計算MATH,所以還需要先計算一個VAF值(t_alt_count/t_depth)
data@data$VAF <- data@data$t_alt_count/data@data$t_depth然后挑一個樣本計算下MATH(這里只用默認參數,具體分析可能會再調試下)
> head(data@data$Tumor_Sample_Barcode) [1] TCGA-D8-A1XY-01A-11D-A14K-09 TCGA-D8-A1XY-01A-11D-A14K-09 [3] TCGA-D8-A1XY-01A-11D-A14K-09 TCGA-D8-A1XY-01A-11D-A14K-09 [5] TCGA-D8-A1XY-01A-11D-A14K-09 TCGA-D8-A1XY-01A-11D-A14K-09 986 Levels: TCGA-3C-AAAU-01A-11D-A41F-09 ... TCGA-Z7-A8R6-01A-11D-A41F-09 res <- inferHeterogeneity(maf = data, tsb = 'TCGA-D8-A1XY-01A-11D-A14K-09', vafCol = 'VAF') > unique(res$clusterData$MATH) [1] 57.76873暫時還沒比較過傳統方法計算的MATH值和maftools包計算的MATH有多少差異。。但是從思路上來看maftools加上聚類的算法去除outline的方法感覺更加合適點?
參考資料:
What does this following maf column mean?
http://bioconductor.org/packages/release/bioc/vignettes/maftools/inst/doc/maftools.html